Válasszon országot vagy régiót


-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)

-
Italia (Italiano)

-
Hungary (Magyar)
-
Lietuva (Lietuviškai)

-
Mexico (Español)

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)

-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)

-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
A kiadványban szereplő személyek valódi betegek, és történetük felhasználásához a betegek illetve családjaik hozzájárultak. A fényképek kizárólag illusztrációként szolgálnak.


MI AZ AZ SMA?
MI OKOZZA AZ SMA-T?
A gerincvelői izomsorvadás (SMA) egy ritka, genetikai eredetű neuromuszkuláris betegség, amelyet a túlélő motoros neuron 1 (survival motor neuron 1, SMN1) gén mutációja okoz.1


MI AZ AZ SMA?
AZ SMA TÍPUSAI
Az SMA különféle típusokba osztható a betegség jelentkezésekor betöltött életkor és a funkcionális képességek szerint. Az egyes típusokon belül pedig több súlyossági fok létezik.2


MI AZ AZ SMA?
HOGYAN ÖRÖKLŐDIK AZ SMA?
Az SMA egy örökletes betegség, és más ritka neuromuszkuláris betegségektől eltérően a gerincvelői izomsorvadás konkrét genetikai okát egyértelműen ismerjük.3
MI AZ AZ SMA?
HASONLÓ TÜNETEKET OKOZÓ BETEGSÉGEK
Habár az SMA bizonyos jelekkel és tünetekkel jellemezhető, vannak más olyan betegségek, amelyek hasonló tüneteket okoznak, ám genetikai okaik különbözőek.

MI AZ AZ SMA?
AZ SMA DIAGNOSZTIZÁLÁSA ÉS VIZSGÁLATA
Amikor az SMA gyanúja felmerül, fontos, hogy a kezdeti diagnózist genetikai vizsgálattal is megerősítsék.4
Mi a Biogennél elköteleztük magunkat a gerincvelői izomsorvadással élők és az ellátásukat végző csoportok támogatása mellett. Reméljük, hogy az „Együtt az SMA-ban” program keretében nyújtott információkkal hozzájárulhatunk ahhoz, hogy Ön a legjobb ellátásban részesüljön, valamint ahhoz, hogy felkészülten tudjon egyeztetni a kezelőorvosával.



MI AZ AZ SMA?
MI OKOZZA AZ SMA-T?
A gerincvelői izomsorvadás (SMA) egy ritka, genetikai eredetű neuromuszkuláris betegség, amelyet a túlélő motoros neuron 1 (survival motor neuron 1, SMN1) gén mutációja okoz.1

MI AZ AZ SMA?
AZ SMA TÍPUSAI
Az SMA különféle típusokra különíthető el a betegség jelentkezésekor betöltött életkor és a funkcionális képességek szerint. Az egyes típusokon belül pedig több súlyossági fok létezik.2

MI AZ AZ SMA?
HOGYAN ÖRÖKLŐDIK AZ SMA?
Az SMA egy örökletes betegség, és más ritka neuromuszkuláris betegségektől eltérően a gerincvelői izomsorvadás konkrét genetikai okát egyértelműen ismerjük.3

MI AZ AZ SMA?
HASONLÓ TÜNETEKET OKOZÓ BETEGSÉGEK
Habár az SMA bizonyos jelekkel és tünetekkel jellemezhető, vannak más olyan betegségek, amelyek hasonló tüneteket okoznak, ám genetikai okaik különbözőek.
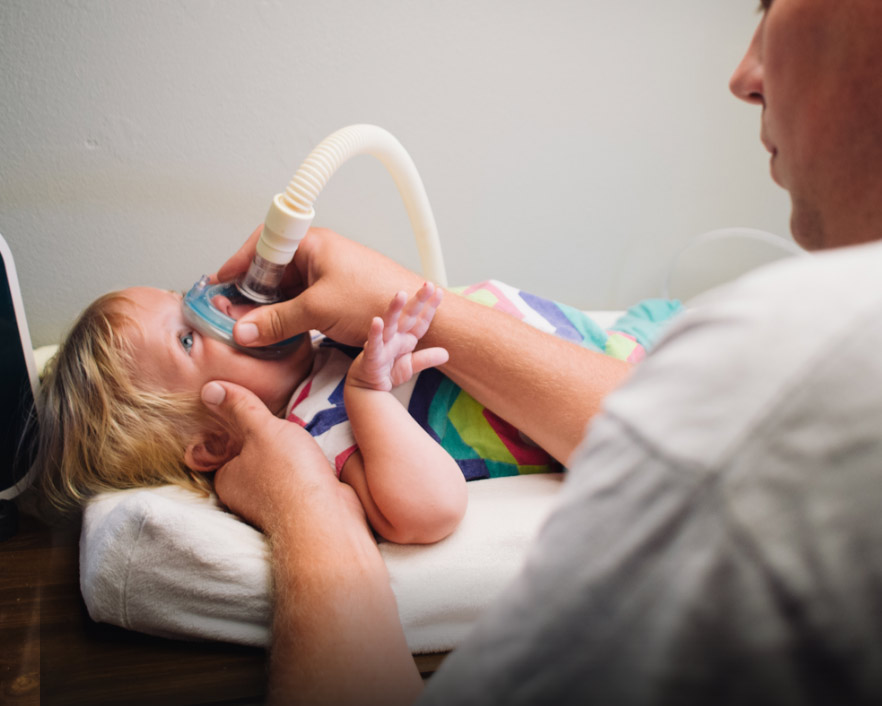
MI AZ AZ SMA?
AZ SMA DIAGNOSZTIZÁLÁSA ÉS VIZSGÁLATA
Amikor az SMA gyanúja felmerül, fontos, hogy a kezdeti diagnózist genetikai vizsgálattal is megerősítsék.4
Mi a Biogennél elköteleztük magunkat a gerincvelői izomsorvadással élők és az ellátásukat végző csoportok támogatása mellett. Reméljük, hogy az „Együtt az SMA-ban” program keretében nyújtott információkkal hozzájárulhatunk ahhoz, hogy Ön a legjobb ellátásban részesüljön, valamint ahhoz, hogy felkészülten tudjon egyeztetni a kezelőorvosával.
A kiadványban szereplő személyek valódi betegek, és történetük felhasználásához a betegek illetve családjaik hozzájárultak.
A fényképek kizárólag illusztrációként szolgálnak.
“Az SMA diagnosztizálásának folyamata nem változott az eredeti konszenzusnyilatkozat óta... hacsak nincsenek korábbi családi esetek, a diagnosztikai folyamatot általában a klinikai jelek indítják el.” 4
Referenciák
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
2. Kolb SJ, Kissel JT. Spinal muscular atrophy. Arch Neurol. 2011;68(8):979-984.
3. National Organization for Rare Disorders. Spinal Muscular Atrophy. [online] 2012 [cited 2020 Sep 22].
Available from: URL: https://rarediseases.org/rare-diseases/spinal-muscular-atrophy/.
4. EGL Genetics. Congenital Hypotonia Panel: Spinal Muscular Atrophy Deletions, Prader-Willi/Angelman Syndrome Methylation, Myotonic Dystrophy, and Uniparental Disomy 14. Available at: http://geneticslab.emory.edu/tests/HY. Accessed January 9, 2017.
5. Mercuri E, et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord 2018;28(2):103-115.