Válasszon országot vagy régiót


-
Argentina (Español)

-
Australia (English)

-
Austria (Deutsch)

-
Bahrain (العربية)

-
Bahrain (English)
-
België (Nederlands)

-
Belgique (Français)
-
Brazil (Portugues)

-
Canada (English)

-
Canada (Français)
-
Chile (Español)

-
Colombia (Español)

-
Croatia (Croatian)

-
Denmark (Danish)

-
Deutschland (Deutsch)

-
Europe (English)

-
France (Français)

-
Greece (Ελληνικά)

-
Italia (Italiano)

-
Hungary (Magyar)
-
Lietuva (Lietuviškai)

-
Mexico (Español)

-
日本 (日本語)

-
대한민국 (한국어)

-
Kuwait (العربية)

-
Kuwait (English)
-
Nederland (Nederlands)

-
Norge (Norsk)

-
Oman (العربية)

-
Oman (English)
-
Polska (Polskie)

-
Portugal (Portuguese)

-
Qatar (العربية)

-
Qatar (English)
-
Saudi Arabia (العربية)

-
Saudi Arabia (English)
-
Slovakia (Slovak)

-
Slovenia (Slovenščina)

-
Spain (Español)

-
Suomi (Suomi)

-
Sverige (Svenska)

-
Schweiz (Deutsch)

-
台灣 (中文)

-
United States (English)

-
UAE (العربية)

-
UAE (English)
MI AZ A GERINCVELŐI IZOMSORVADÁS (SMA)?
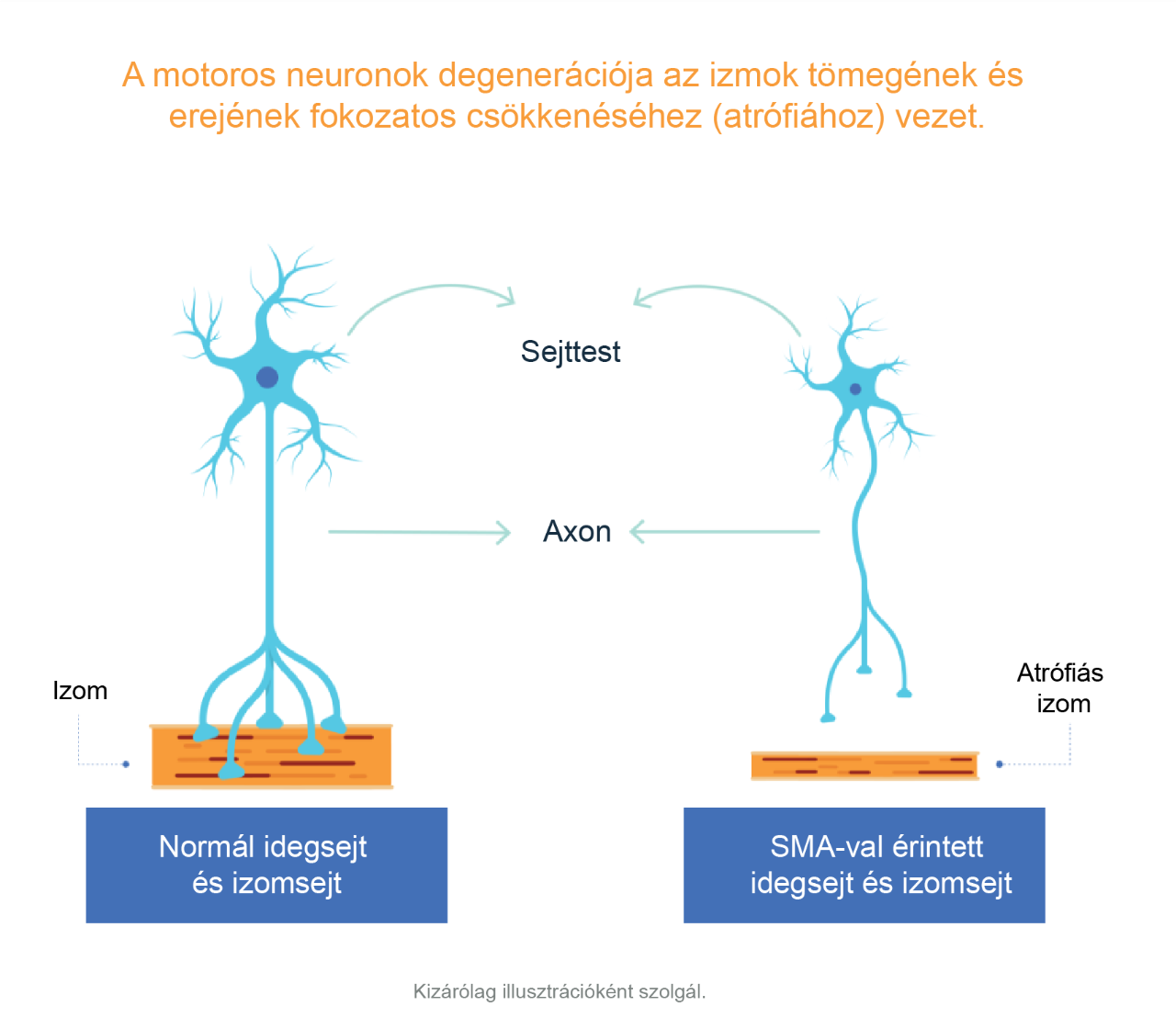
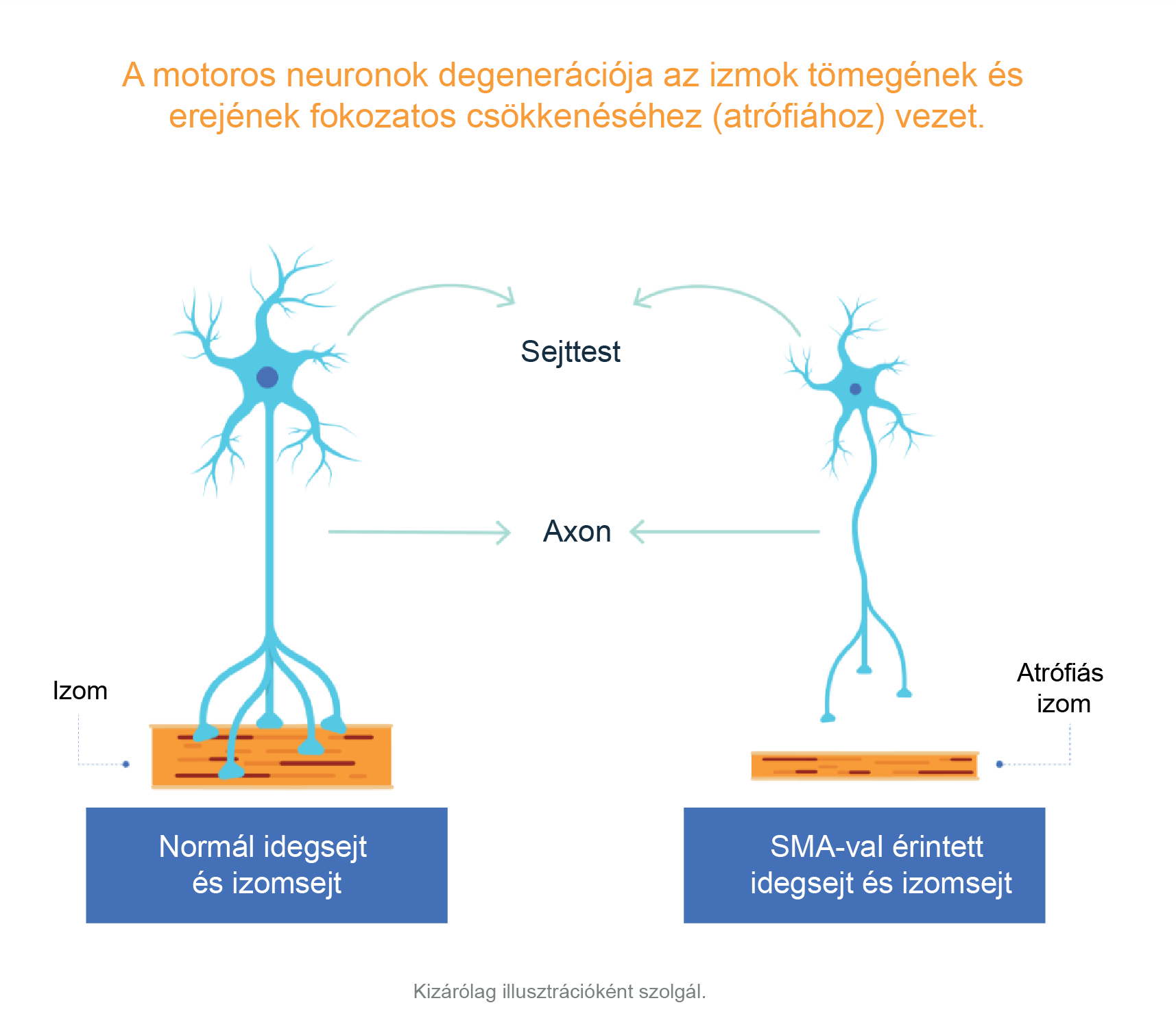
Az SMA az idegrendszernek az akaratlagos izommozgást irányító részét érinti.1 Gerincvelői izomsorvadásban a gerincvelő azon fontos sejtjei – a motoros neuronok –, amelyek létfontosságúak az izmok erejének megőrzéséhez és azok mozgatásához, elpusztulnak. Ezek a motoros neuronok azáltal szabályozzák az izomaktivitást, hogy jeleket küldenek a központi idegrendszerből (KIR), azaz az idegrendszernek az agyat és a gerincvelőt magában foglaló részéből.1,2
A motoros neuronok funkciójának elvesztése fokozódó izomgyengeséghez és atrófiához (az izomtömeg és az izomerő fokozatos csökkenéséhez) vezet, mivel az izmok többé nem kapnak jeleket a KIR-ből.3
A GERINCVELŐI IZOMSORVADÁS OKÁNAK MEGÉRTÉSE
Más ritka neuromuszkuláris betegségektől eltérően a gerincvelői izomsorvadás konkrét genetikai okát egyértelműen ismerjük. Az SMA-t a túlélő motoros neuron 1 (SMN1) génben bekövetkező mutáció okozza, amely gén a túlélő motoros neuron (SMN) fehérje termelődéséért felelős. Ez a fehérje fenntartja a motoros neuronok egészségét és normál működését.
A gerincvelői izomsorvadásban szenvedő emberekben az SMN1 gén mindkét kópiájában mutáció történt – ez pedig az SMN-fehérje csökkent termelődését eredményezi. Megfelelő mennyiségű SMN-fehérje hiányában a gerincvelőben található motoros neuronok idővel elpusztulnak, ami miatt az izmok nem lesznek képesek megfelelő jeleket fogadni az agyból.4
Itt többet is megtudhat a gerincvelői izomsorvadás öröklődéséről
Az SMA különféle típusokba osztható a betegség jelentkezésekor betöltött életkor és a funkcionális képességek szerint. Az egyes típusokon belül pedig több súlyossági fok létezik.5


CSATLAKOZZON
és tudjon meg többet arról, hogyan lehet része az SMA közösségnek
A kiadványban szereplő személyek valódi betegek, és történetük felhasználásához a betegek illetve családjaik hozzájárultak. A fényképek kizárólag illusztrációként szolgálnak.
Referenciák
1. Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
2.Wang CH, et al. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-1049.
3. National Institute of Neurological Disorders and Stroke. Motor Neuron Disease Fact Sheet. 2012.
Available at: https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Motor-Neuron-Diseases-Fact-Sheet. Accessed January 9, 2017.
4. Genetics Home Reference. SMN2 gene. 2012. Available at: https://ghr.nlm.nih.gov/gene/SMN2. Accessed January 9, 2017.
5. Kolb SJ, Kissel JT. Spinal muscular atrophy. Arch Neurol. 2011;68(8):979-984.